Close menu
-
Windows Apps
-
Wordpress
-
Linux Apps
-
MAC Apps
0 Reviews
FREE
License
PAID
Version
LatestUpdate
Last updated
OS
Language
EN

WH was developed to be a computer program that carries out the fitting of a speciation model, and conducts tests of the quality of fit of that model. The speciation model is called the Isolation Model, and is one without gene flow. With comparative DNA sequence data from each of two closely related species, the method allows an estimation of the time since speciation and the size of the ancestral species.
The isolation model assumes the following: - Two species, or populations, from which the data have been sampled, arose from a single ancestral species, or population, some time, t generations, in the past. - The common ancestral species, or population was of constant effective size NA. - The two descendant species (populations) have constant effective sizes N1 and N2, respectively. - Except for the moment of population separation at time t, population sizes are constant. - Since population separation, there is zero gene flow between the species (populations). - All mutations are neutral.
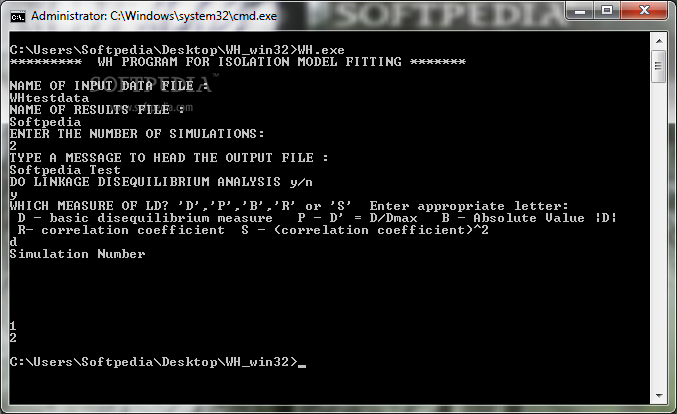
The program file should reside either in the same folder as the data file or in a folder automatically searched by the operating system. The program can be run using command line parameters, or by simply typing the name of the program ('wh'). If command line parameters are not used, the program asks for the values of runtime parameters.
The user starts the program simply by going to the folder where the data file exists and typing the name of the program (e.g. 'sites') followed by the enter key. The program asks several questions about the data file and the desired analysis. Nearly all commands and options can also be entered using command line parameters.
The program can be started with or without the use of instructions at the command line. Without command line instructions - simply type ‘wh’ at the prompt. The program will ask for basic information.
On a PowerPC, clicking on the program icon opens a small window in which command line parameters can be entered. The user can also just hit return at this point and the program will request runtime parameters.
Command Line Parameters:
Type and enter
wh -d'datafilename' -r'resultsfilename' -N'numsims' -L'ldtype' -A'ranseed'
Where:
'datafilename' is the name of a data file that is in the same folder as the program
'resultsfilename' is the name of the file that will be created to contain the results. The results file will be generated with an extension of '.wh'
'numsims' is an integer of the the number of simulations to be run. Basic model fitting does not require simulations. If this value is not zero or blank, the data file must have the necessary information on recombination rates.
'ldtype' tells the program what type of linkage disequilibrium value to calculate. This option requires that simulations be done, and that the data file include information on recombination and observed LD values. 'ldtype' must correspond to the measure of linkage disequilibrium used to generate the datafile.
'ldtype' can take on the following values: 'R' invokes the correlation coefficient 'S' invokes r^2, the square of the correlation coefficient 'D' invokes the basic gametic disequilibrium measure D 'P' invokes the D' which is equal to D/ Dmax, where Dmax is the maximum possible value of D given the observed allele frequencies. 'B' invokes the absolute value of D, |D|
'ranseed' is a random number seed and is not generally required unless the user wants to repeat simulations exactly.
No reviews found
SoftPas is a platform that provides you with the latest software and technology news, reviews, and guides. We also provide you with the latest software and technology trends.
Subscribe to newsletter
© Copyright 2024, All Rights Reserved by SoftPas